5. PRIMER PROJECTS
- 5.1 primer design
- 5.1.1 allowed characters
- 5.1.2 syntax check
- 5.1.3 primer and file name
- 5.1.4 how to design primers
- 5.1.5 functions
- 5.1.6 back-translate protein sequence
- 5.2 primer properties
- 5.3 back-translate protein sequence
- 5.4 primer header
- 5.4.1 primer file name
- 5.4.2 primer sequence information line
- 5.4.3 long primer name
- 5.4.4 chemical modifications of oligonucleotides
- 5.5 order primers by e-mail
- 5.5.1 primer order form
- 5.5.2 information retrieved from header
- 5.5.3 order text
- 5.5.4 save options
- 5.5.5 print options
- 5.5.6 view order file
- 5.5.7 sending order by e-mail
- 5.5.8 layout of e-mail order form
- 5.6 order form viewer
- 5.7 order preferences
 5.1 primer design
5.1 primer design
This function File/Enter Sequences Manually/Primer Sequences is used to construct new primers for PCR amplification and DNA sequencing. When this option is selected the sequence export format is set to primer. Note that a new file must be created for each primer. The right mouse button activates a popup menu including common options for primer design.
If a primer sequence is loaded as the FIRST sequence of a normal sequence project
the project type is automatically set to primer design mode and subsequent files to be loaded
must all be in primer format or be converted to this format.
Using SEQtools as a primer "database" including the primer sequences in a normal nucleotide project offers several advantages: The primer sequences can be annotated/verified by Genbank searching relevant databases. This annotation is stored in the sequence header of the primer sequence. You can then use sequence listing and searching facilities to identify for example primers for a particular organism, gene etc.
5.1.1 Allowed characters
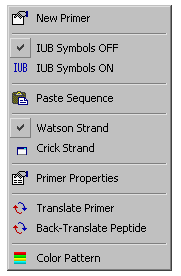
Clicking the Format button, formats the primer sequence with a block length of 3 and a line length of 30. It also adds the symbols
- X = modified position
- I = Inosin
- ( = left parenthesis
- ) = right parenthesis
as well as the IUB symbols for degenerate base positions to the list of valid characters. The position counter in the lower right of the editor window displays the actual number of bases in the primer - counting degenerate positions as one.
5.1.2 Syntax check
Degenerate base positions are either written in full or abbreviated by using IUB symbols. During formatting the primer sequence, syntax checking is performed. A warning which includes some common errors is issued if the primer sequence contains one or more of the following syntax errors.
- - Empty brackets ( )
- - Unpaired brackets ( (
- - Identical bases in brackets (AA)
- - A single base in brackets (A)
- - 3' position is degenerate ACCTGT(ATG)
5.1.3 Primer and file name
The default name and extension of primer files is PRIMER.001 . The primer file names can be batch-changed with the file name template (Edit/Names/Edit Sequence Names) or change name functions. Specific, long primer names of up to 60 characters can be entered in the header form accessed from Header/Primer Header. If the user fails to enter a long primer name the primer file name is used instead.
5.1.4 How to design primers - a brief description
- - Click File/Enter Sequences Manually/Primer Sequencesto open a new project.
- - Enter the sequence of the primer in the empty file.
- - Click Update in the main editor to accept and format the sequence.
- - Right mouse button to open the popup menu and click Primer Properties to display the primer data.
- - On popup menu click IUB Symbols ON/OFF to toggle between normal and IUB symbols display.
- - Change the default file name PRIMER.001 to a unique file name in the main editor.
- - Click Header / Primer Header in the main editor to open the primer header.
- - In the primer header, enter the name of the primer, select modifications and user comments.
- - On popup menu click New Primer to create a new empty file for the next primer.
- When finished, click File/Save or Export Files to store project.
5.1.5 Functions, etc.
Default format - The default primer formatting is line length of 30 characters with blocks of 3.
Copy and paste primers - Primer sequences can be pasted into an empty file from other applications including second instances of SEQtools.
Convert to complementary strand - Click Edit/Crick Strand on the main editor form or on the popup menu to convert the primer sequence to its complement.
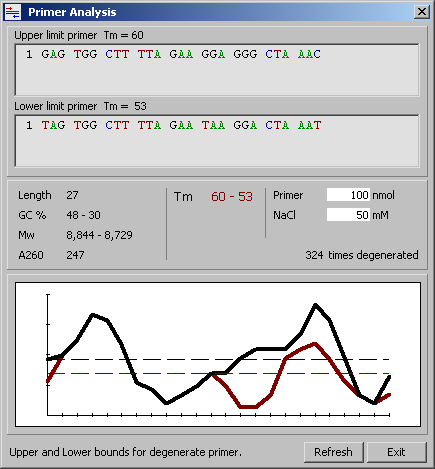
Primer properties - The primer properties can be displayed by clicking Analyse/Primer Properties on the main editor form or on the popup menu. If the primer includes degenerate positions the melting profile is calculated for the degenerate primer sequences with the lowest and the highest melting points.
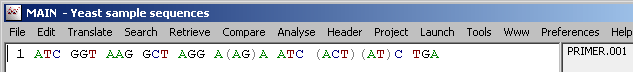
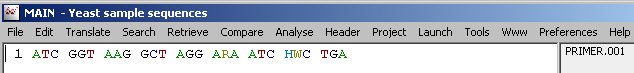
Degenerate positions - Degenerate positions in the primer sequence are either included in parentheses (IUB Symbols OFF) or typed using the IUB Symbols as illustrated by the two figures below:
Degenerate positions by full base annotation:
Degenerate positions by IUB symbols:
5.1.6 Back translate protein sequence
Click Translate / Back-Translate Peptide on the main editor menu to
back-translate an amino acid sequence into a degenerate primer. Before you can use this
option you must type or copy/paste an amino acid sequece into an empty primer file.
Also note that is necessary to select and load a codon usage file to provide relevant codon
usage information for back-translation before this function can be used. You will be prompted
to do so when you attempt to back-translate an amino acid sequence. SEQtools automatically
prompts you to load either a *.cod (IUB format) or *.cut (seqtools format) file.
5.2 primer properties
This form Analysis/Primer Properties shows several characteristics of the current primer sequence including the number of bases, the GC percentage, the molecular weight, the A260 value and the Tm.
5.2.1 Melting temperature, Tm
The melting temperature, Tm value is calculated according to the nearest-neighbor method using the delta-H, delta-S and delta-G values from Breslauer et al. (1986). Proc. Natl. Acad. Sci. USA, 83, 3746-3750.
If the sequence contains inosins, I, or modified positions, X, the mean delta-H, delta-S and delta-G values are used for the di-nucleotides including the I's or X's and the melting profile is displayed in red. For normal primers, the melting profile is shown in black. The melting profile is displayed graphically, averaged over four bases along the sequence.
For primers with degenerate positions, the primer properties function determines the primer sequences with the smallest and largest Tm value and displays the two sequences with the minimum and maximum Tm for the degenerate primer sequence. An example is given below illustrating the function.
5.2.2 Calculation of Tm value for the sample primer
(
A
T
G
)
A
G
T
G
G
C
T
T
T
T
A
G
A
A
(
T
G
A
)
(
T
G
A
)
A
G
G
(
T
A
G
)
C
T
A
A
A
(
A
T
C
G
)
The calculation of the two Tm values for the lower and upper limit sequence is based on the following di-nucleotides ranked as:
ranking = -deltaH/(-deltaS -13.909)
DiBaseRanking( 1) = "TA" ' deltaH = 7.2 deltaS = 21.3 ranking = 0.204
DiBaseRanking( 2) = "AT" ' deltaH = 7.2 deltaS = 20.4 ranking = 0.210
DiBaseRanking( 3) = "AA" ' deltaH = 7.9 deltaS = 22.2 ranking = 0.219
DiBaseRanking( 4) = "TT" ' deltaH = 7.9 deltaS = 22.2 ranking = 0.219
DiBaseRanking( 5) = "AG" ' deltaH = 7.8 deltaS = 21.0 ranking = 0.223
DiBaseRanking( 6) = "CT" ' deltaH = 7.8 deltaS = 21.0 ranking = 0.223
DiBaseRanking( 7) = "GA" ' deltaH = 8.2 deltaS = 22.2 ranking = 0.227
DiBaseRanking( 8) = "TC" ' deltaH = 8.2 deltaS = 22.2 ranking = 0.227
DiBaseRanking( 9) = "AC" ' deltaH = 8.4 deltaS = 22.4 ranking = 0.231
DiBaseRanking(10) = "GT" ' deltaH = 8.4 deltaS = 22.4 ranking = 0.231
DiBaseRanking(11) = "CA" ' deltaH = 8.5 deltaS = 22.7 ranking = 0.232
DiBaseRanking(12) = "TG" ' deltaH = 8.5 deltaS = 22.7 ranking = 0.232
DiBaseRanking(13) = "CC" ' deltaH = 8.0 deltaS = 19.9 ranking = 0.237
DiBaseRanking(14) = "GG" ' deltaH = 8.0 deltaS = 19.9 ranking = 0.237
DiBaseRanking(15) = "GC" ' deltaH = 9.8 deltaS = 24.4 ranking = 0.256
DiBaseRanking(16) = "CG" ' deltaH =10.6 deltaS = 27.2 ranking = 0.258
5.2.3 Primer properties for a primer with degenerate positions
This form displays a number of parameters for the primer sequence. For primers with degenerate positions the parameters for the primer sequence with the highest and the lowest Tm value are calculated and displayed. Click the Refresh button to update the properties after editorial changes of the primer sequence.
5.3 back-translate proteiN sequence
This function Protein/Back-Translate Protein performs a back-translation of a peptide sequence into a degenerate DNA sequence and calculates the degree of degeneration. If the degeneration exceedes 10,000 times, the actual value is not displayed.
5.3.1 Validation of sequence
Be careful not to confuse a primer sequence written with IUB symbols and a peptide sequence. The program will issue a warning if the peptide sequence appears invalid (i.e., appears to be a DNA sequence including IUB symbols).
5.3.2 Codon usage tables
Before back-translating an protein sequence a codon usage table must be loaded
using the File menu on the codon usage form. SEQtools accepts two types of tables,
either one generated and modified by the user *.cut or species specific codon usage tables
imported in IUB format, *.cod. The latter type of tables can be converted to SEQtools format
by simply by saving them with the *.cut extension whereas it is NOT possible to save codon
usage files in IUB format *.cod.
Seqtools prompts you to select and load a codon usage table when you try to back-translate a
peptide sequence. To accept the loaded codon usage table simply close the codon tabel
editor after the table is successfully loaded into the editor. The selected table will be
active until you change it by loading a new table (Analysis/Codon Usage).
5.3.3 Degeneration level
The degree of degeneration of primers created by back-translation can be controlled by selecting a degeneration level between 1 and 6, where 1 implies that only the preferred codons are used for back translation (and the resulting primer is without degenerate positions). Choosing degeneration level 6 means that maximum degeneration is achieved and all possibilities are covered in the primer. Values between 1 and 6 uses the 2, 3, 4 or 5 most frequent codons for each amino acid. Obviously this will increase the degeneracy of the primer, only if more than one codons encodes a particular amino acid.
5.3.4 Strand
The peptide sequence can either be back-translated into a forward primer or a reverse primer. In the latter case, the back-translated sequence is converted into the complementary strand before the calculation of the degree of degeneration.
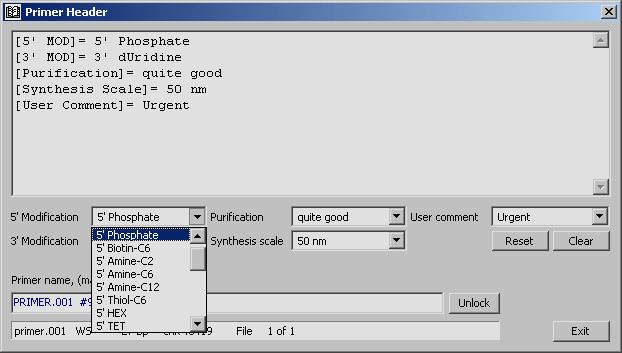
5.4 primer header
The primer header allows you to create and edit a sequence-specific header including
various types of information related to the primer currently displayed in the editor. The entire
content of the primer header including all field is attached to the header of the current primer
when the file is saved.
5.4.1 Primer file name
The primer file name can be edited manually in the main seqtools form and batchwise with the special functions for renaming files found under the Edit menu (Edit/Names/Edit Sequence Names).
5.4.2 Primer sequence information line
This line lists the file name, orientation of the sequence and the checksum of the displayed primer sequence.
5.4.3 Long primer name
The current file name for the sequence is displayed when the header form is opened. Long primer names up to 60 characters in length can be entered in this field and will be attached to the file header. The normal file/sequence name used for saving the sequences will remain unaffected by editing the long primer name. Note that you must unlock this field before the long primer name can be edited. The normal primer file/project name is local and will not be displayed in the primer ordering form.
5.4.4 Chemical modifications of primers
Four dropdown boxes are included in the primer header form to enable you to specify various 3' and 5' modifications, a preferred purification method and a user comment to your primer sequence.
Selecting a modification for either (or both) ends of the primer causes a field to be added to the header text: [3' MOD]= for 3' modifications and [5' MOD]= for 5' prime. The values of these fields are added to the primer order. The text can be selected from the dropdown lists, entered manually in the text field of the dropdown list or in the header text itself. Notice that the maximum length for all four manual entry types is limited to 40 characters. Include here production related information relevant for the primer, eg. purification method, coupling, origin, etc.
5.5 order primers by e-mail
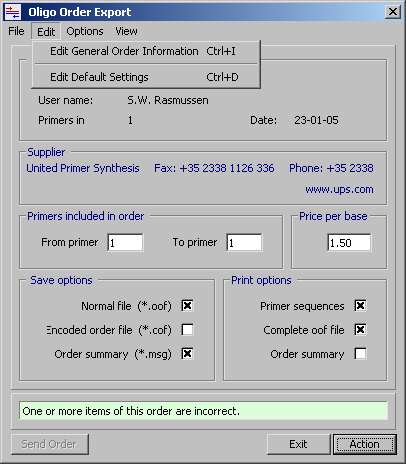
This form includes facilities for constructing, saving, printing and e-mailing primer order forms. Select the operations you want to perform and press Action. The selected save and print jobs will then be carried out sequentially. If the price per base in entered in the price text field, the order summary includes a price calculation for each length group of primers. Note that is not possible to e-mail the order before it is saved.
5.5.1 Primer order form
This form is used to compose the order form to be sent by e-mail to
the oligonucleotide supplier. Before the form is usable the various standard indormation for
the specified supplier must be entered in Preferences/Primer Ordering Settings
as described in section 5.7.
5.5.2 Information retrieved from the header
The primer export order function automatically retrieves the following information from the primer header:
- - Long primer name
- - 5' modifications of the primer ([5' MOD]= entry)
- - 3' modifications of the primer ([3' MOD]= entry)
- - The first 80 characters of header text (only if this option is selected in Order Text)
5.5.3 Order text
General information for the primer order is entered by clicking this button and filling in the necessary information if the different text fields. The Remarks text box includes three options for including sequence specific remarks.
When order verification if on, the order text is veryfied and the user warned if information is missing or incorrect. Items in the verification list marked with a star should be checked before shipping the order. Turning verification off cancels this check.
5.5.4 Save options
This section of the export form contains three check boxes for selecting save items:
- - A normal primer order file, file extension: *.oof
- - An encoded primer order file, file extension: *.cof
- - A summary of the order, file extension: *.msg
Click the check boxes for the operations you want to carry out and press the Action button. The selected save jobs will then be carried out sequentially.
When any of the three check boxes are checked, the user is prompted for a file name for the primer order file. The name of the three files will be the same with the file-specific extensions added. If the file already exists, the user is warned before overwriting.
Before the order files are saved, a sequence specific checksum is generated and saved with each primer sequence. The calculation of the checksum for the primer sequence is the same as that used by SEQtools for normal sequence but is done on the formatted sequence also taking spaces between blocks into account.
If the Encoded order file box is checked you will be prompted to enter - and reenter - the pincode of max 25 characters to be used by the encoding algorithm. Press <ENTER> after each entry of the pincode. In case the two entries differ, the user is warned and must re-enter the pincode. The same pincode is required for decoding the order file. Note that you cannot create an encrypted order file without first generating a normal order file.
A simple XOR algorithm is used for encoding the order file which only provides limited protection against unautherised access to the information contained in the file. Generally a long pincode (max 25 characters) makes the file more difficult to decode. Obviously the receiver must have access to the pincode used to encode the file.
If a price per base in entered in the price text field, the order summary includes a price calculation for each length group of primers.
The format of a normal order file is shown below. Note that each field of each record uses one line. Records are delimited by a /SOR/.../EOR/ pair.
5.5.5 Print options
This section of the export form contains three check boxes for selecting print items:
- - The primer sequences
- - The complete, normal order file
- - A summary of the order
Click the check boxes for the operations you want to carry out and press the Action button. The selected save jobs will then be carried out sequentially. The layout for the primer sequence print is shown below. The complete order file and the order summary are printed exactly as they are saved.
5.5.6 View Order
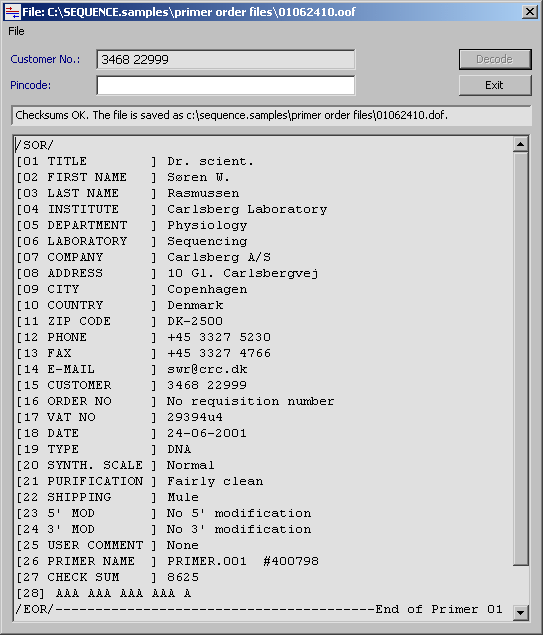
Clicking this command button displays the primer order viewer (Tools/File Tools/View Primer Order) which enables you to view both normal order files, encrypted order files and the mail.log file. Simply open the file you wish to view and follow the instructions on the form. You need to enter the pincode before an encrypted order file can be decoded and displayed. The order text is displayed exactly as it appears on the saved *.oof and *.cof. The decode function automatically saves a copy of the order file with the extension *.dof.
5.5.7 Sending the order by e-mail
When the order form is correctly filled out, the order can be sent automatically from SEQtools by clicking the Mail button. It is possible to enter a message to the supplier.
5.5.8 Layout of the e-mail order form
Each primer of an order file comprises a RECORD. Each record contains 27
single-line text fields delimited by /SOR/ and /EOR/ as shown below.
Start of user message
/SOM/
User message, inserted in front of order file if the file is sent by the e-mail function. This message is not included saved *.oof or *.ocf file.
/EOM/
End of user message
Start of primer record
/SOR/
[01 TITLE ] Entered in order text as described above
[02 FIRST NAME ] Entered in order text
[03 LAST NAME ] Entered in order text
[04 INSTITUTE ] Entered in order text
[05 DEPARTMENT ] Entered in order text
[06 LABORATORY ] Entered in order text
[07 COMPANY ] Entered in order text
[08 ADDRESS ] Entered in order text
[09 CITY ] Entered in order text
[10 COUNTRY ] Entered in order text
[11 ZIP CODE ] Entered in order text
[12 PHONE ] Entered in order text
[13 FAX ] Entered in order text
[14 E-MAIL ] Entered in order text
[15 CUSTOMER NO ] Entered in order text
[16 ORDER NO ] Required, entered in order text
[17 VAT NO ] Entered in order text
[18 DATE ] Current system date
[19 TYPE ] From preferences/order text
[20 SCALE ] From preferences/order text
[21 PURIFICATION ] From primer header or From preferences/order text
[22 SHIPPING ] From preferences/order text
[23 5' MOD ] From primer header, value of [5' MOD]= in header text
[24 3' MOD ] From primer header, value of [3' MOD]= in header text
[25 PRIMER NAME ] From primer header
[26 USER COMMENT ] From primer header, value of [User comment]=
[27 CHECKSUM ] Check sum is recalculated by the decode function)
[28 SEQUENCE ] Sequence in upper case, block of 3, GCG symbols)
/EOR/
End of primer record
5.6 order form
Viewer
This utility (Tools/File Tools/View Primer Order) is used to decode encoded oligo primer orders *.cof and to view normal, un-encoded order files *.oof. To decode encoded order files, use the same pincode as was used when the order file was encoded. The file is marked read only and the text cannot be edited in this viewer.
Use the page up and page down keys to navigate between individual records of the oligo primer order. The decoded file is automatically saved in the directory from which is was loaded and gets the same name with the extension *.dof , (decoded oligo order file).
5.6.1 Checksum verification
After decoding the order file, all primer specific checksums are recalculated
and the user notified if there are any discrepancies between the original and the
recalculated checksums.
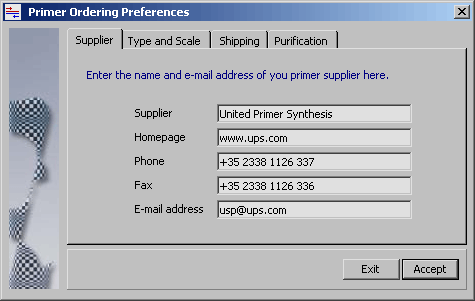
5.7 order preferences
The four tabs on this form (Preferences/Primer Ordering Settings) contain
various text fields to customise the order text in the primer ordering form to fit the
requirements of different suppliers. The content entered in the text field serves as default
values in the primer order text form.
If for example the purication method has been
specified in the header of a particular primer, this value overrides the default. The tabs
include the following information:
© 2002-2010S.W. Rasmussen (revised: )