4.6 COMPARE MENU
- 4.6.1 about comparing sequences
- 4.6.2 compare two sequences (general)
- 4.6.3 compare two sequences (dot matrix)
- 4.6.4 compare two sequences (bl2seq)
- 4.6.5 alignment, clustalw
- 4.6.6 alignment, clustalx
- 4.6.7 sequence merge editor
- 4.6.8 merge sequences, emboss
- 4.6.9 clustering
 4.6.1 about comparing sequences
4.6.1 about comparing sequences
This menu contains a series of functions for comparing and/or aligning two or more sequences.
Alignment is performed by ClustalW for functions with a SEQtools user interface. It is possible to use the facilities
contained in the ClustalX version of the alignment program which comes with its own user interface - with many more
options than contained in the SEQtools functions.
SEQtools communicates to a limited extent with ClustalX (auto-exporting sequences to ClustalX and importing the
alignment when processing is completed and ClustalX is closed).
4.6.2 compare two sequences (general)
This compare function performs a comparison between two sequences selected from the project sequence list.
If no sequence is selected the currently displayed sequence is compared with itself.
The result is displayed as
a sorted list of matches on the current sequence giving the length of identical regions and the coordinates of the regions
on the current and the query sequence. A W indicates the match is to the Watson strand and a C a
match to the complementary Crick strand (in red). Note that coordinates refer to the Watson strand of both
the current and the query strand. Various parameters (max number and min length of matches) can be set
on the Options menu.
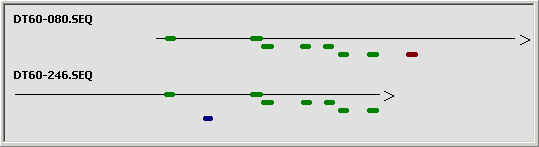
To get a quick overview matching (=identical) regions between the two sequences are displayed in a diagram
with identical regions indicated in green (W strand) or blue (C strand).
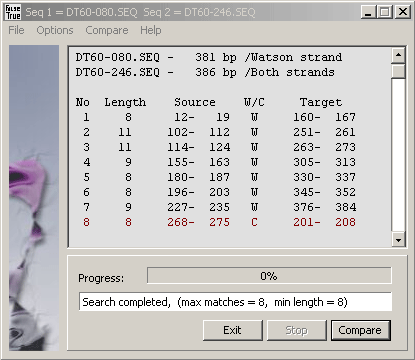
Graphical overview of the sequence comparison for identical regions.
4.6.3 compare two sequences (dot matrix)
This function allows you to compare two sequences (nucleotide or protein), or a sequence with itself,
using a dot matrix approach for finding identical regions in the two sequences (selected from the project sequence list)
by clicking the Select command button.
A matrix is built by clicking Build containing boolean information (True/False) for the comparison of all bases in
sequence 1 against all bases in sequence 2. When the matrix is complete click Filter to clean (of short
diagonals) and display the matrix. Clicking Filter again increments the minimum length of preserved
matches. You can manually enter a value in the text field before clicking Filter to achieve the same result.
The dot plot is scaleable.
Holding down the left mouse button while searching the dot plot with the mouse pointer, displays the coordinates in
both sequences. Releasing the mouse button snaps to the nearest diagonal (if you are sufficiently close to it) and
displays the properties of the match (diagonal).
For protein sequence comparisons it is possible to set and select a replacement scheme which causes the
function to replace single character amino acid codes by numbers representing amino acids with similar
properties.
The different options of the dot plat function is illustrated by screenshots below. All examples shows a comparison
of a single sequence with itself.
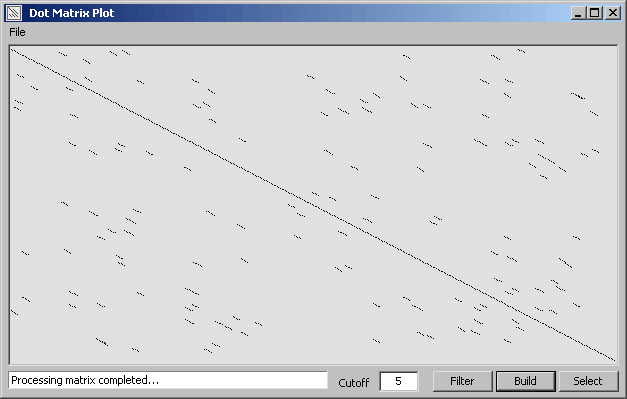
A short nucleotide diagonal detected by pointing and dragging while holding down the left mouse button.
The diagonal is auto-detected when the left mouse button is released.
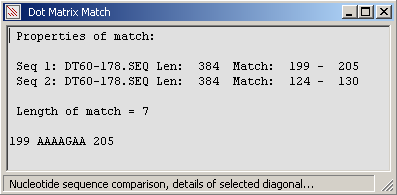
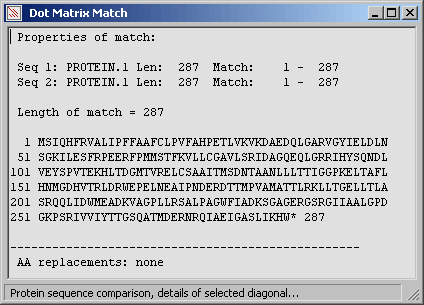
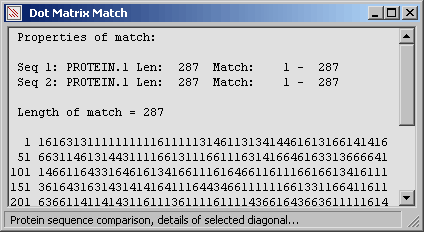
The full diagonal (perfect match of the sequence to itself) of the comparison.
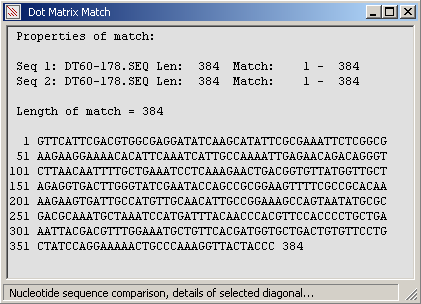
The amino acid replacement form for composing replacement schemes. You can create and
store upto 10 different replacement schemes. Click Acccept to use the selected scheme - and Build to
see the result of the amino acid replacement.
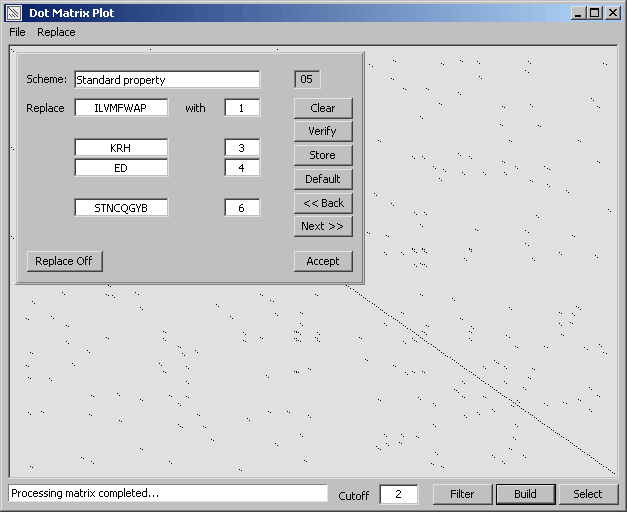
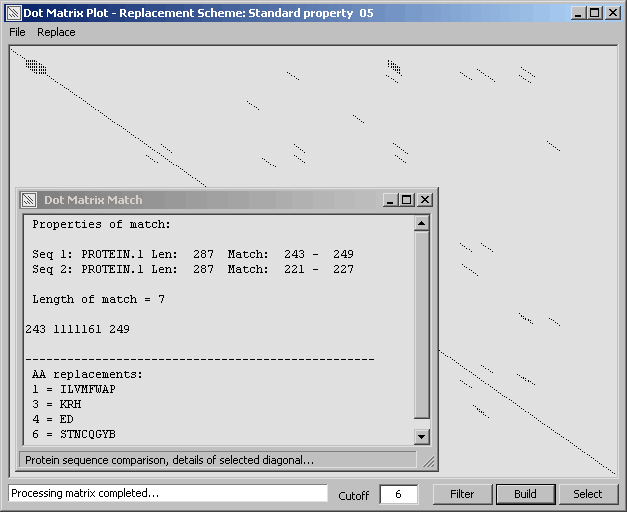
Dot plot of an amino acid sequence compared to itself created with amino acid replacement activated. - The
diagonal analysis window includes coordinates to the match as well as the replacement scheme.
The longest diagonal from the comparison above without amino acid replacement.
The same diagonal but with active amino acid replacement to highlight regions composed of amino
acids with similar properties.
4.6.4 compare two sequences (bl2seq)
This function utilising the NCBI program bl2seq allows you to compare the currently displayed sequence
to other sequences contained in the project. The actual comparison is performed by blastn, blastx or blastp depending on
the program argument of bl2seq. The SEQtools interface to bl2seq auto-detects the sequence type to be
compared in the following way:
The Subject sequence is for protein and nucleotide projects the currently selected/displayed nucleotide sequence.
Query sequences are either the currently selected nucleotide sequence (in Single Search mode) or a selected range
of nucleotide sequences included in the project (Project Search mode).
Query = nucleotide, Sbjct = nucleotide, Program = blastn - Default for normal sequence projects.
Query = protein, Sbjct = protein, Program = blastp - Default for protein projects. Options controlling
upper/lower/both query strand(s) are disabled in this mode.
Query = nucleotide, Sbjct = protein, Program = blastx - Activated nucleotide projects when the current sequence is translated into protein. The displayed translated sequence (i.e., a protein sequence) is retrieved directly from the main
editor and used as the subject in the
comparison with bl2seq.
Default blast arguments:
Default settings for bl2seq
Relaxed blast arguments:
Cost to open a gap = 0 Cost to extend a gap = 0 Dropout value for gapped alignment = 0 Word size = 8 Penalty for a nucleotide mismatch = -1 Reward for a nucleotide match = 2 Other bl2seq parameters = defaults for bl2seq
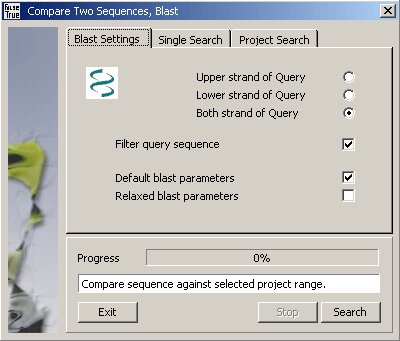
Use the Strand options to compare the upper, lower or both strand of the Query sequence(s) against
the Subject sequence.
The output is stored in the sequence header of the query sequence(s). Note that you can only store the results of one
comparison analysis at a time. Running a new bl2seq analysis overwrites existing data stored in the headers
without warning.
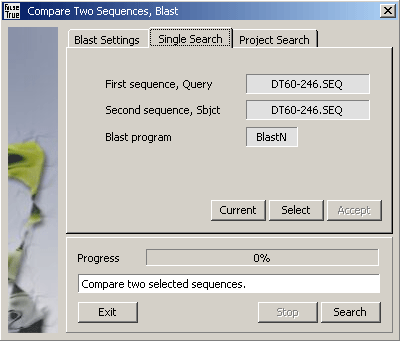
Single bl2seq comparison of two selected sequences.
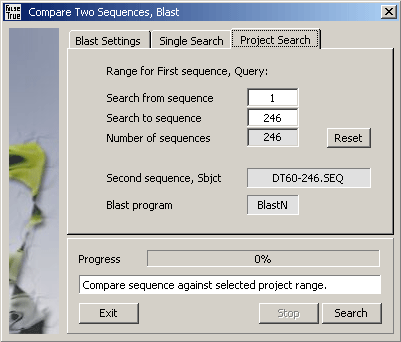
Comparing the Subject sequence to all or a selected discontinuous range of project sequences.
4.6.5 alignment, clustalw
, Muscle
This function uses the Clustal programs - or the recently added Muscle program to perform the actual alignment. The ClustalW and the Muscle programs ares
fully integrated into SEQtools and it is not possible to change the default parameters used to for the
alignment. ClustalX (described in the following section of the manual) is a stand-alone Windows program with
its own user interface including a large number of options influencing the alignment proces. The File menu of
the SEQtools alignment form allows you to choose which of the two programs you wish to use.
The SEQtools interface to the fully integrated ClustalW program allows you to specify the output format and - on the second
tab of the alignment form to alter the sequence names used by ClustalW in the finished alignment. These options are not available for the Muscle program. The Select command
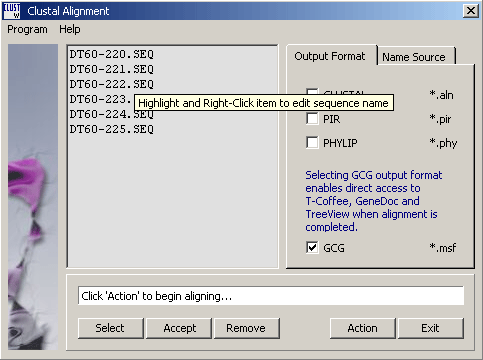
button opens the project sequence list for selection of sequences to align:
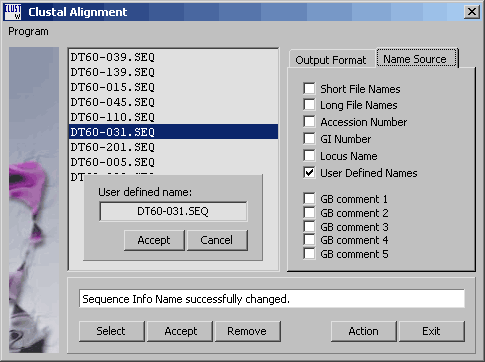
Clicking the Accept command button, populates the sequence list with the selected sequences. You can edit the list only by removing iteme (select and click Remove). If you wish to post-process the alingment, choose the GCG output format to enable the menu options on the alignment result form to the GeneDoc, TreeView and T-Coffee programs provided GeneDoc and TreeView are properly installed on your pc and thet SEQtools has been informed by adding the two programs to the Launch menu. Clicking Action starts the alignment process displaying the progress in the info line of the form.
The tab for selecting a different source for the names for the sequences to be aligned. Shift-Right clicking the sequence list opens a smal text editor to allow you to edit individual sequence names one at a time.
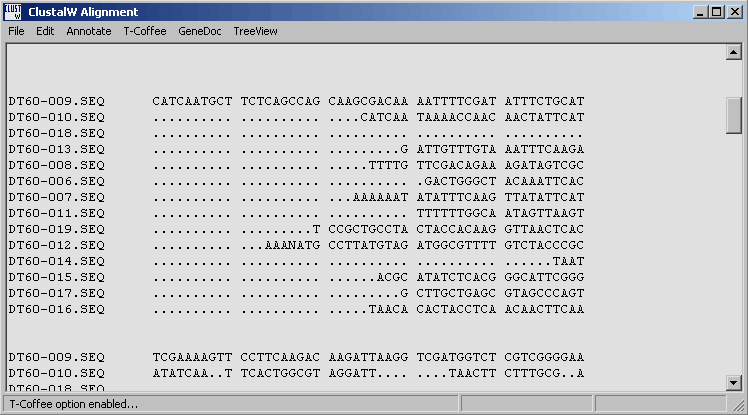
The output form of a Clustal and Muscle alignment. Note that the menu line contains direct access to the programs GeneDoc, TreeView and T-Coffee.
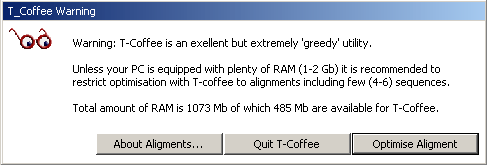
4.6.5.1 T-Coffee - is a very powerful routine for optimising the initial clustal alignment. Users with sufficient RAM (in most cases t-coffee requires 0.5 - 1.0 Gb of RAM if you want to optimise alignments consisting of more that a few sequences). SQTtools warns you in the message displayed below also informing you of the amount of free RAM on you pc. Optimisation wil terminate if the pc runs out of ram while optimising the alignment.
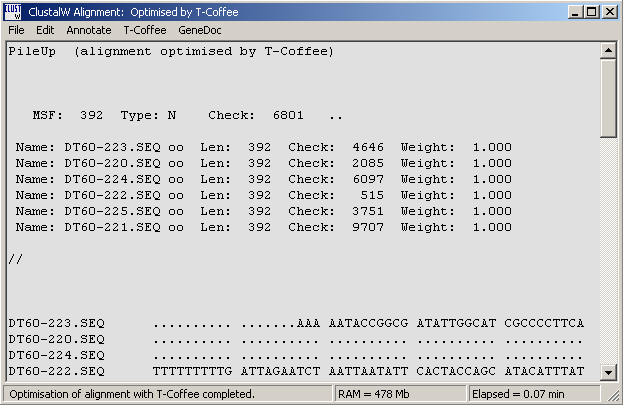
Successful optimisation with T-Coffee is indicated in the top line of the alignment as shown below.
4.6.5.2 GeneDoc - is an extremely powerful alignment editor providing facilities for very advanced editing and annotation of the alignment.
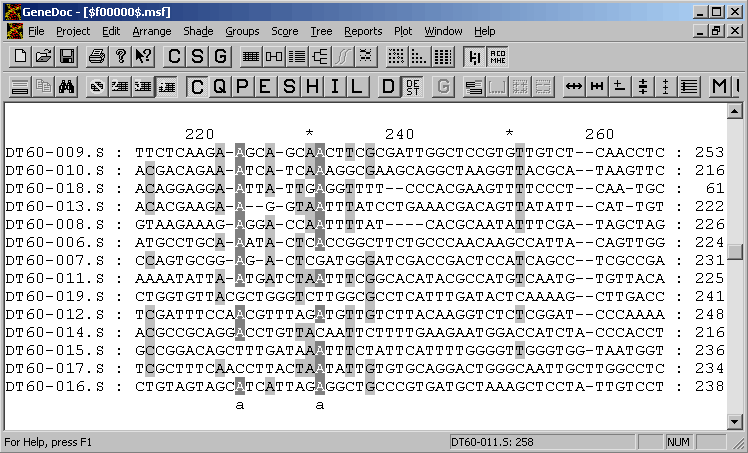
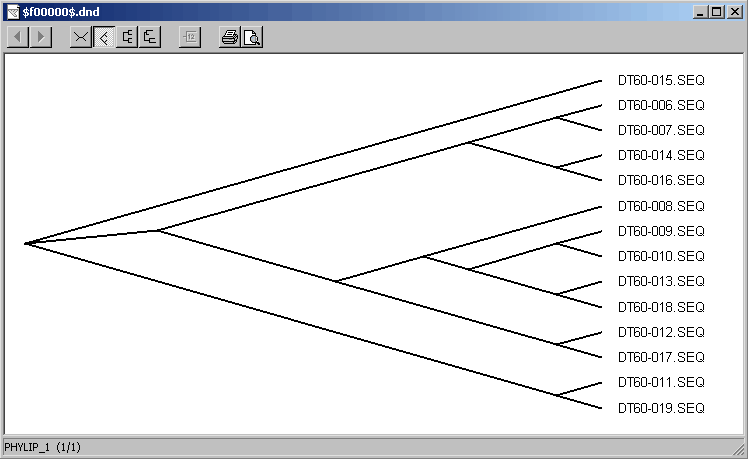
4.6.5.3 TreeView - this small program enables you to build a phylogenetic tree of the aligned sequences at a single click.
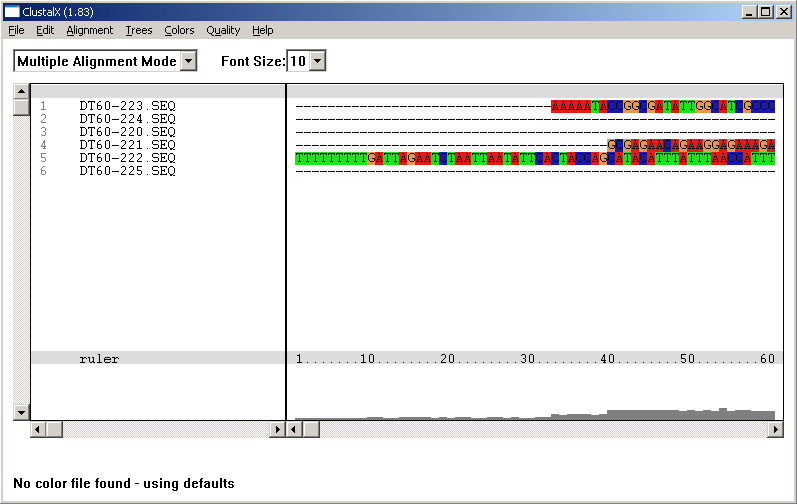
4.6.6 alignment, clustalx
The ClustalX program is powered by the same routine as ClustalW but has its own user interface allowing you to specify a large number of parameters for the alignment. The resulting file - when saved in GCG format - is auto-imported into SEQtools when you close ClustalX.
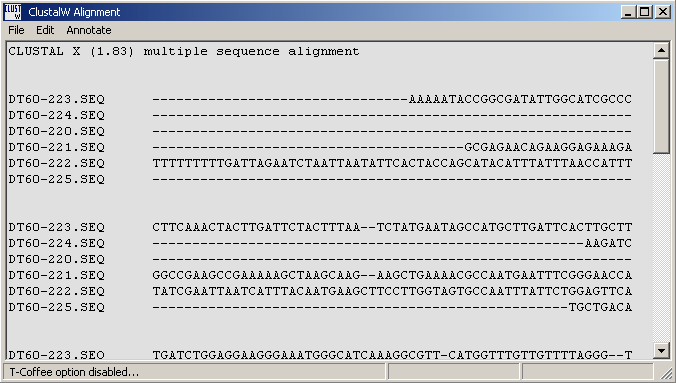
Alignment created by ClustalX and re-imported into SEQtools after closing ClustalX.
4.6.7 sequence merge editor
This function is designed to assist you in building and editing merges of overlapping nucleotide sequences.
Although this merge function appears rather primitive, extended use of the function for finishing small sequencing projects
has demonstrated that it works quite effectively in creating an error-free consensus sequence from the sub sequences.
Click Select to display the project sequence list for selecting the sequences to me merged - when done click Accept.
Set the Preferences (main menu) and click Merge. It is not possible to change preferences after
the sequences are merged so be sure to do it correctly before you click Merge.
When merging is complete a small window pops up informing you about included and excluded sequences, the page number and the
bases on the page.
The merged sequences are contained on separate, fixed pages. You navigate between the pages of the merge by the <Page UP> and <Page Down> keys. Press the >CTRL + G> keys to display the goto field and the <ENTER> key to jump to the specified base - or to close the goto field.
The bottom panel on the merge editor informs you about sequence name of the clicked sequence, total length and number of discrepancies (in red) on the displayed page.
When editing is completed, click the Verify command button to rebuild the merge and the consus sequence
displayed above the merge. This enables the Save and Print options under the File menu. You can
append the consensus sequence to the current project by clicking Build/Store Consensus and Build/Update
Project to update the project sequences with the changes made during editing the merge.
Remember that all information is SEQtools is stored in RAM om your pc until the project is saved. To undo
the changes made while editing the merge, simply close the project without saving it.
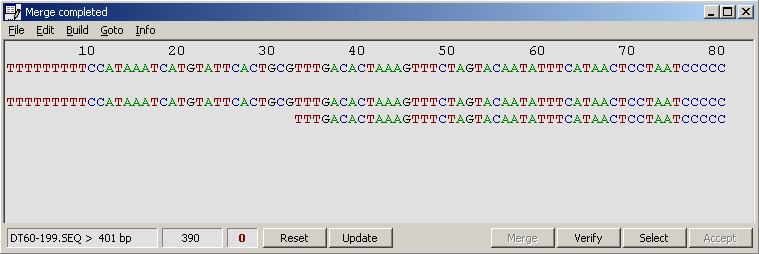
The merge editor info form.
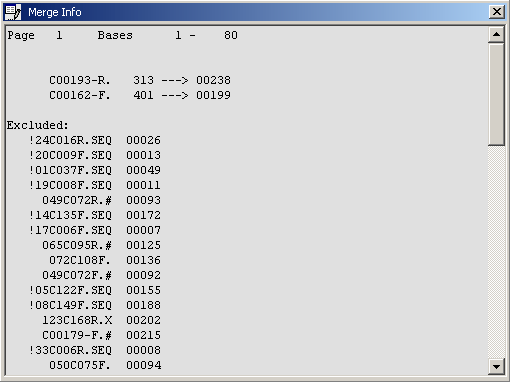
Setting the preferences for the merge editor must be set before merging the selected sequences.
4.6.8 merge sequences, emboss
A small feature which was used to automatically merge forward and reverse sequence for EST
inserts on the same plasmid.
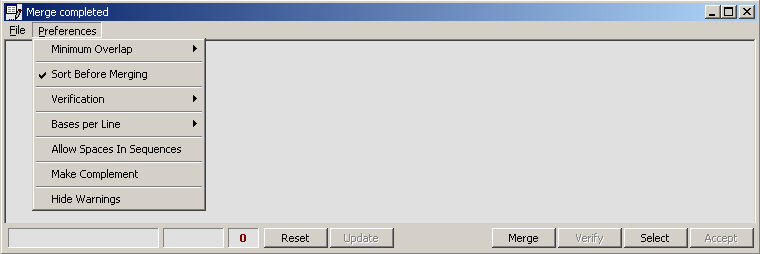
The function requires that clones are named as indicated on the merge form: forward and reverse sequence must have
identical leftmost characters (first six characters of the sequence name) and a closing -F
for forward and -R for reverse sequences of the same insert. The merged sequence will receive -M as
the last two characters of their sequence name.
The merged sequences are appended to the current project.
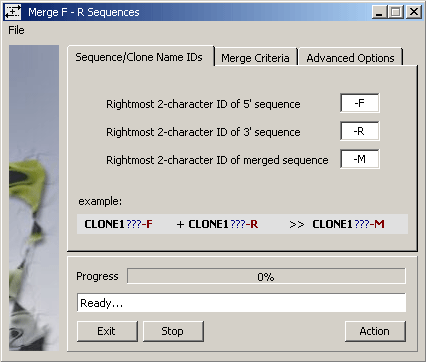
The SEQtools function uses the emboss Merger routine to perform the actual
merging with the argument options shown on the Merge Criteria tab of the form (below).
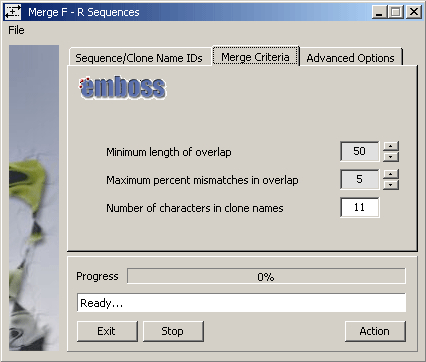
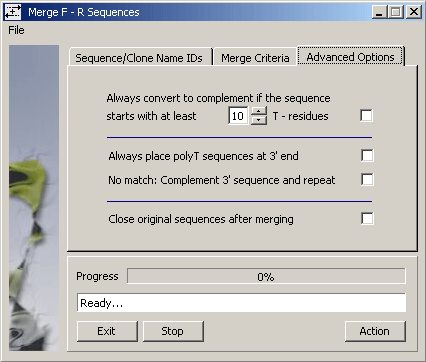
The Advanced Options tab enables you to tell SEQtools to complement sequences to be
merged, to close (i.e., remove from current project) the -F and -R sequences successful merged.
4.6.9 clustering
This function allows you to perform nucleotide sequence clustering with three different methods:
- based
on score values,
- by the NCBI blastclust program
- by pairwise sequence alignments.
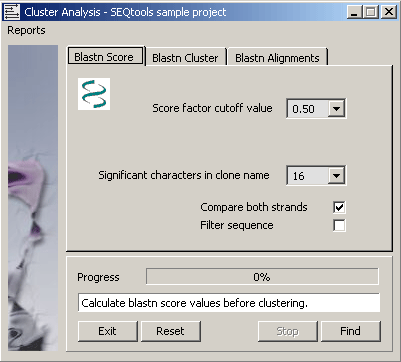
4.6.9.1 Blast score values - The function automatically creates a local database with formatdb (NCBI) and
performs the blastn search against this to generate temporary blastn score values used for clustering. The self-score value
(the score value obtained when the sequence is compared to itself) is extracted for all sequences contained in the project.
Then, the score value for each local blastn match is normalised by division with the self-score value. Finally, the normalised
score values are compared to the selected score-cutoff limit and matches less than the cutoff are rejected.
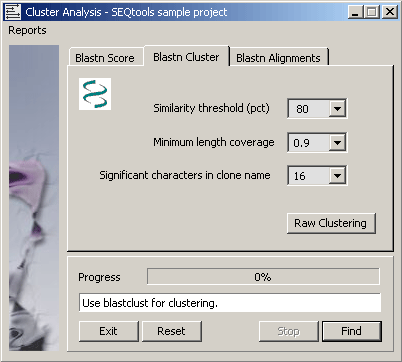
4.6.9.2 Blastclust program - Blastclust automatically and systematically clusters protein or DNA sequences
based on pairwise matches found using the blast algorithm in case of proteins or mega blast algorithm for DNA. In the
latter case a single Mega BLAST search is performed for all the sequences combined against a database created from the
same sequences. Blastclust finds pairs of sequences that have statistically significant matches and clusters them using
single-linkage clustering.
Blastclust uses the default values for the blast and mega blast parameters. For protein sequences these are: matrix
Blossum62; gap opening cost 11; gap extension cost 1; no low-complexity filtering. For DNA sequences: match reward 1,
mismatch penalty -3, non-affine gapping costs, wordsize 28. In both cases e-value threshold is set to 1e-6.
(from the NCBI blastclust documentation).
4.6.9.3 Blastn alignments - This function first creates a local database including all sequences in the project
with formatdb, performs a blastn search for all sequences and collects the best alignment for each sequence. The
alignments are parsed to identify sequence pairs sharing a minimum length identical region with the specified number of
mismatches. The list is used to generate the reports described below.
The three functions will in most cases yield the same overall clustering of sequences. There are, however, situations where
the blastn alignment-based method yields a more reliable clustering: Long sequences with small overlaps from the same
open reading frame may be lost when blastn clustering is used but will, if they share just a short identical region be
linked by the alignment method. On the other hand, sequences from different ORFs sharing a short identical region may
erroneously be linked with the identical region method but not when local blastn score data are used.
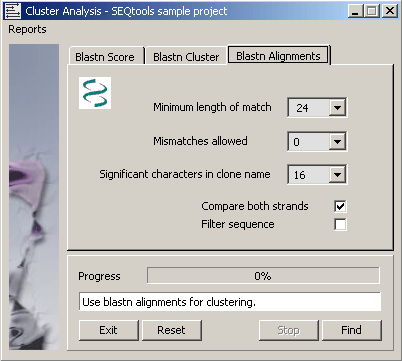
4.6.9.4 Output - A cluster tree listing all clusters resulting from the clustering process. Clicking a node
in the tree retrieves a list of description lines for the cluster. As for the project sequence list, selecting a line and
holding down the right mouse button in the cluster member list displays the blast descriptions for the selected sequence.
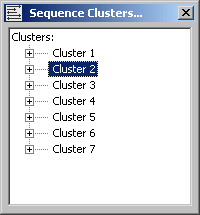
The cluster group form. Note that there are direct access (when two or more members of the cluster group are highlighted) to Aligning and Merging (in the merge editor described above)the members of the cluster group on the menu.
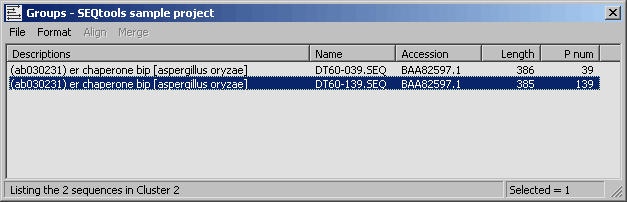
4.6.9.5 Clustering report - The clustering report simply lists the clusters generated by either method. A
summary section is included in front of the list giving overall values for redundancy, number of unique clones in the
project etc.
4.6.9.6 Deduced clone report - The deduced clone report processes the sequence report by removing all but
one of sequences sharing the specified number of file name characters. This option allows you to remove multiple
sequences originating from the same clone provided that the sequences are named consistently:
Example:
original cluster:
CLONE001-2.SEQ CLONE001-4.SEQ CLONE001-1.SEQ CLONE002-2.SEQ CLONE002-3.SEQ CLONE003-7.SEQ CLONE003-8.SEQ CLONE005-4.SEQ CLONE005-12.SEQ
processed cluster (significant characters = 9):
CLONE001-2.SEQ CLONE002-2.SEQ CLONE003-7.SEQ CLONE005-4.SEQ
The two optional reports, File/Sequence Cluster Report and the File/Deduced Clone Report
are displayed on a separate Result form.
© 2002-2010S.W. Rasmussen (revised: )