4.3 TRANSLATE MENU
- 4.3.1 about translating nucleotide sequences
- 4.3.2 translate in specified frame
- 4.3.3 find in all frames
- 4.3.4 translate forward frames
- 4.3.4.1 file menu (translate form)
- 4.3.4.2 view menu (translate form)
- 4.3.4.3 frame menu (translate form)
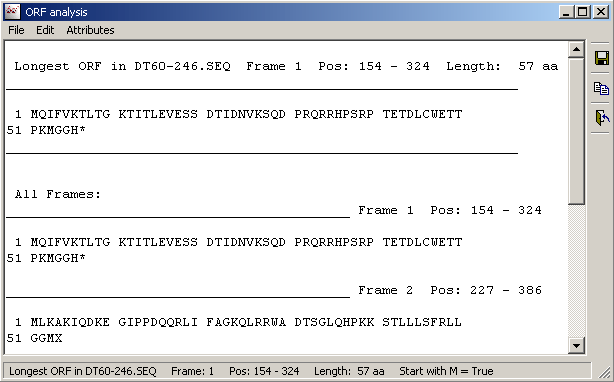
- 4.3.4.4 format menu (translate form)
- 4.3.4.5 attribute menu (translate form)
- 4.3.4.6 transfer selected region (from translate form)
- 4.3.5 back-translate protein sequence
- 4.3.6 create protein files
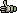 4.3.1 about translating nucleotide sequences
4.3.1 about translating nucleotide sequences
SEQtools includes several options for translating nucleotide sequences as well as for back-translating protein sequences. With the Find In All Frames is is possible to identify the longest ORF in a nucleotide sequence. The Translate Specific Frame you can isolate (Copy/Paste) the isolated protein sequence). The Translate Forward Frames provide a link between the translated sequence and the underlying nucleutide sequence. Finally it possible to batch translate all nucleotide sequences contained in the project with the Create Protein Files function.
![]()
4.3.2 translate in specified frame
This function enables you to translate a nucleotide sequence in the specified reading frame. You have the
option to display either the longest ORF, the longest frament or a complete translation in the specifed frame.
4.3.2.1 Largest ORF submenu.
![]()
4.3.2.2 Largest Fragment submenu.
![]()
4.3.2.3 Complete Translation submenu.
![]()
4.3.3 find in all frames
With this function it is possible easily to identify the longest ORF (open reading framing) or fragment (protein region without stop codons) in an unknown nucleotide sequence. The result is displayed in text form which lists the longest orfs/fragments in all six reading frames and the longest of them all.
With information you can re-translate the longest ORF/fragment with the Translate Specific Frame
function described above to isolate the protein sequence.
![]()
The Result form listing ORF's or fragments in all six readinf frames of the nucleotide sequence.
4.3.4 translate forward frames
The Translate Forward Frames displays the translation of the current DNA sequence or an extract
thereof in each of the three forward reading frames or in all three forward reading frames simultaneously.
The line numbers correspond to the coordinates of the extracted sequence region. Stop codons are denoted by stars and
uncertain (codons including one or more N's) amino acids by X's. The format of the DNA sequence is independent of the
selected format in the sequence editor form with block length of 3 and line length of 60 bp.
4.3.4.1 File menu - contains save and prints options for the translated sequence.

4.3.4.2 View menu - includes the available translate options.

4.3.4.3 Frame menu - selects the reading frame (forward only) for the translation.

4.3.4.4 Format menu - allow you to select line length and whether or not to divide the sequence in blocks of 10 residues.
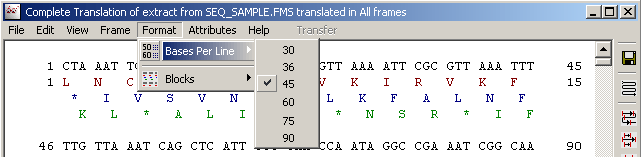
4.3.4.5 Attributes menu - contains simple options for annotating the translated nucleotide sequence.
4.3.4.6 Transfer - it is possible with the transfer options to highlight a nucleotide region (for example corresponding to an interesting portion of the translation and - by clicking Transfer - to transfer the highlights to the normal sequence editon (see below).
![]()
Highlights corresponding to the selected region in the Forward Frame Translation form.
![]()
Re-translating the highlighted nucleotide region in the normal sequence editor displays the translation with the translated nucleutides displayed above the protein sequence.
![]()
4.3.5 back-translate protein sequence
Back-translating protein sequences is useful when designing sequencing primers. When a protein sequence is displayed in the normal sequence editor selecting the Back-Translate option prompts you to select/load a codon usage data file to supply information about frequently used codons (codon usage) for the particular organism/protein.
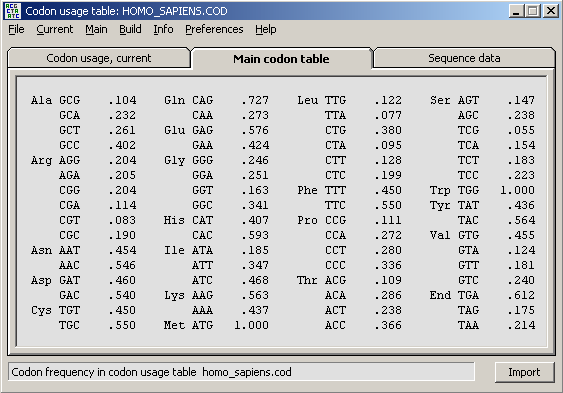
When a codon usage data file is successfully loaded into SEQtools the form below is displayed to enable you to select a degeneration level. Choosing leve1 1 will result in a primer without degenerate positions while level 6 will cover all possible degenerate base combinations. The cost in the latter case of course is few primers in the mixture with the correct base sequence exactly matching the nucleotide sequence.
![]()
The primer sequence after back-translation is displayed in a simple text form. You must then copy/paste the primer sequence into a separate instance of SEQtools opened for handling primers.
![]()
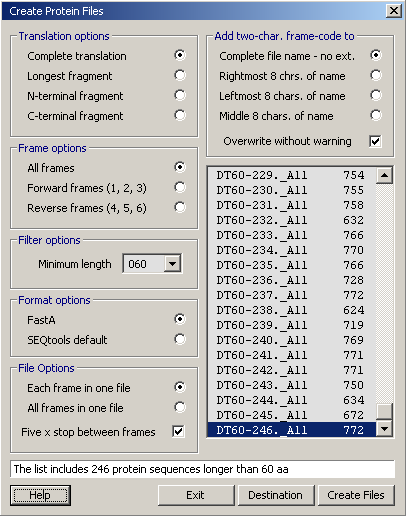
4.3.6 create protein files
This utility is designed to assist you in the analysis of short EST sequences (expressed sequence tags) in cases where functional identification by data base searching has failed and the correct reading frame thus is unknown.
The utility translates all nucleotide sequences of the current project in the selected reading frame(s) and saves each protein sequence in a separate file. The extracted protein sequences can then be searched for example against the Prosite data base of protein motifs, or other data bases including protein signatures.
Translation options:
Complete sequences - the complete translation including X 's and stops.
Largest fragments - largest contiguous amino acid region without stops
N-terminal regions; regions starting with a M and ending at the first downstream stop
C-terminal regions; regions from the start of the sequence to the first stop
Frame options - (1) All reading frames. (2) The 3 forward reading frames (A, B, C). (3) The 3 reverse reading frames (D, E, F).
Filter option - allows you to disregard protein sequences shorter then the selected minimum length.
Protein file names -
The protein file names are constructed by adding _N to the file names of the DNA sequences, where N denotes the reading frame (1-6, or # for all reading frames in the same file). The two characters can be added in one of four ways:
(1) By replacing the extension of the DNA sequence file name with _N. (2) By adding _N to the leftmost six characters of the file name. (3) By adding _N to the rightmost six characters of the file name. (4) By adding _N to the middle six characters of the file name.
In the latter three cases, the protein file names will lack an extension. When the protein files are build, the selected file names are validated to avoid duplicate file names.
If the selected naming method yields duplicate names, the building is arrested and the used advised to select another method of generating protein file names.
In cases where none of the available four methods yields unique protein file names, the original DNA sequence files must be renamed.
File format - The protein files can be saved in either Fasta or GCG format. Each protein file includes a header giving the sequence name, the reading frame and the length of the protein sequence. The protein sequences are broken into lines of 50 characters without line numbering.
Save options - The protein files can either be saved in separate files or in one file per DNA sequence. If the latter option is selected, a 5 x stop separator is inserted between each reading frame if the check box for this option is checked.
© 2002-2010S.W. Rasmussen (revised: )