4.2 EDIT MENU
- 4.2.1 about functions for editing (general comments)
- 4.2.2 undo / redo changes
- 4.2.3 numbering sequence residues (offset and reverse)
- 4.2.4 renaming sequences (compose new sequence names)
- 4.2.4.1 change names
- 4.2.4.2 view renamed sequences
- 4.2.4.3 modify sequence names
- 4.2.4.4 view modified names
- 4.2.4.5 replace I
- 4.2.4.6 replace II
- 4.2.4.7 file and folder tools
- 4.2.5 complementing sequences (complement and invert)
- 4.2.6 trim raw sequences (batch edit sequences)
- 4.2.6.1 remove poly-a tails
- 4.2.6.2 remove vector sequence
- 4.2.6.3 remove low quality sequence
- 4.2.6.4 simple trimming
- 4.2.7 edit project composition (edit project composition)
- 4.2.7.1 remove low quality sequences
- 4.2.7.2 similarity analysis
- 4.2.7.3 remove short / long sequences
- 4.2.8 cut / copy / paste
- 4.2.9 show chromatogram (using Chromas)
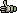 4.2.1 about functions for editing
4.2.1 about functions for editing
Under the Edit menu is collected several functions all directed towards batch editing sequences and
their names. Some are trraight forward others more complex. Below each menu item is explained in some detail.
4.2.2 undo / redo changes
These options allow you to undo editorial changes. Note that changes are not recorded until you press the Update command button in the main editor.
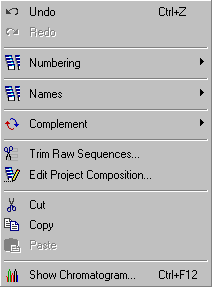
4.2.3 numbering sequence residues
Simply enter a positive or negative value to offset the sequence numbering. Entre a zero to get the normal
numbering back.
4.2.4 renaming sequences
Manual editing of individual file names can be performed by clicking the field displaying the current sequence name on
the main editor form. Editing the names of individual files should be done after batch-renaming all files of the
project.
Batch-renaming will irreversibly eliminate any changes previously made to the names of individual files. The options
for batch editing sequence names are quite complex allowing you to change/edit/customise names in almost any way you can imagine.
On the last tab of this form you can inspect the changes before you implement them by clicking the Apply command
button..
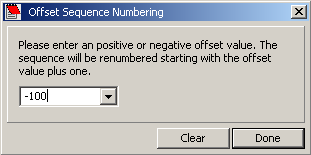
4.2.1.1 Change names - With this function entirely new sequence names can be generated based on a template of 16 characters (the
maximum lenght of sequence names in SEQtools). Type the characters you wish including numerical characters. In the latter case a
check box appears above the character field. Putting a check mark in one or more of the check boxes creates a counter
which will increment by one per sequence.
The example below includes two counters, a 4-digit and a 3-digit counter. See the result of the renaming operation below.
Note that at least one of the counters must be able to hold the total number of sequences in the project.
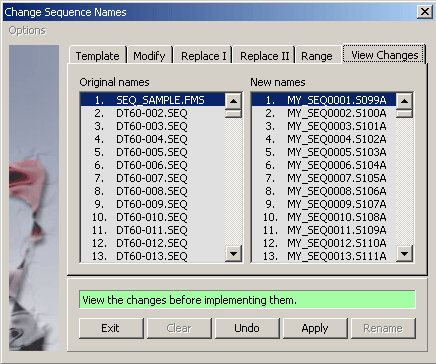
4.2.4.2 View renamed sequences - View the changed sequence names on the panel to the right before implementing the
names by pressing the Apply command button.
4.2.4.3 Modify sequence names - This function makes it possible to make complex changes to parts of the file names without
affecting other parts of the names. The function initially separates the sequence name into the title and extension
and treats the two components of the file name independently.
With this function, characters can be replaced or removed inside the name. Addition or replacement can be made from the left
or from the right of the two parts of the name. The last tab on the form lists the original and the new names of all files
of the project. With this function parts of the old file names can be preserved while unwanted characters can be removed.
The new file names are validated and renaming disrupted if the renaming results in duplicate file names.
Clicking the Apply command button activates the renaming of all files of the project according to the settings of the
options and text. If the renaming operation generates duplicate file names, the operation is interrupted and the remaining
original names are preserved.
Clicking Undo eliminates all changes to the file names of the project. This does not affect changes made to the
sequences and their headers.
The Close command button closes the window preserving the current changes as listed in the new names combo box.
To cancel without renaming, reset the sequence names before closing.
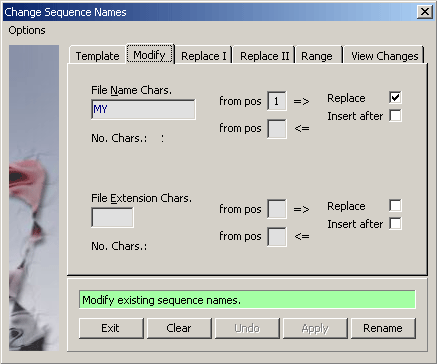
File name characters - This text field can hold up to 8 characters which can
be added to or inserted into the current file names as selected by the options buttons.
Extension characters - This text field can hold up to 3 characters which can be added to or inserted into the
current file extension as selected by the options buttons.
Position fields - The values entered in these fields give the position of insertion or replacements from left or
right of names and extensions.
Add / Insert - This option causes the characters in the text/extension fields to be added/inserted into the file
names/extensions at the position from the left/right as set by the two position fields. Inserting spaces into file
names/extensions has no effect on the file names or extensions.
If the number of characters to be added causes the length of the name plus extension to exceed a total length of 16 characters
the excess characters are truncated from the left or right end of the names and extensions.
Replace - This option causes the characters in the text/extension fields to replace the same number of
characters from the left or right of the file names/extensions as set in the position fields. Replacing characters with
spaces deletes the characters from names/extensions.
Increment - If the text boxes only contain numerical characters a check box appears which, when checked,
causes the increment of the value in the text boxes (increment is one per sequence of the project).
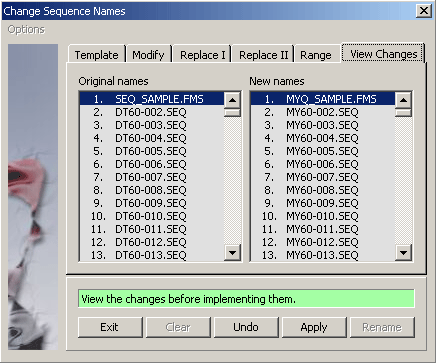
4.2.4.4 View modified names - View the changed sequence names on the panel to the right before implementing the
names by pressing the Apply command button.
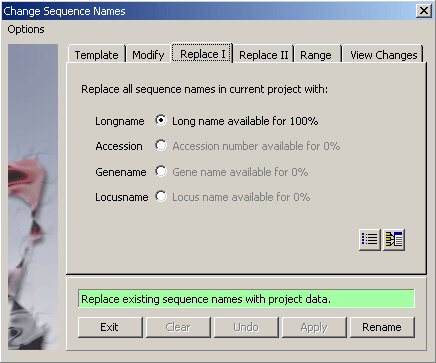
4.2.4.5 Replace I - Batch replace sequence names with one of the enabled categories on this tab. Disabled
options imply that the relevant information is not available for all sequences of the project.
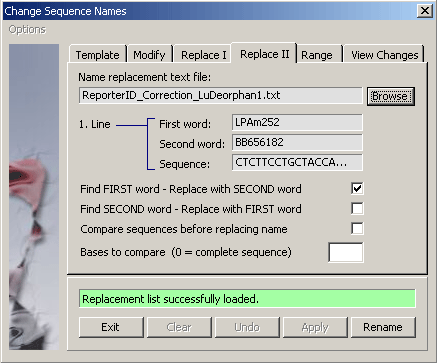
4.2.4.6 Replace II - Complex function to replace project sequence names with the names for the same
sequence but contained in a text file with differens annotation. Eksample: Assume you have an annotated project and a fasta
file with the same sequences. With this function you can replace the project sequence name with the first or the second
word of the fasta definition line. Before replacement takes place the two sequences are compared and only identical sequences
will be renamed.
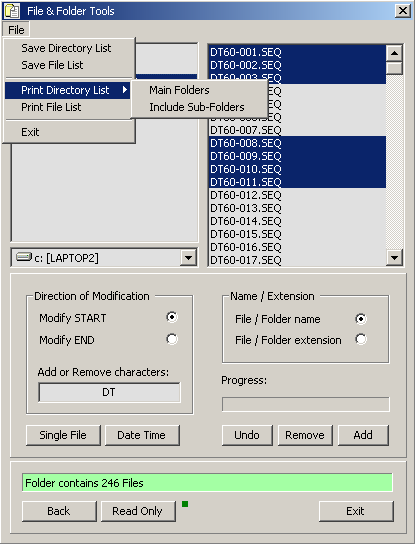
4.2.4.7 File and Folder Tools - This small program enables you to carry out a number of operations on file
and folder names. You can edit the file titles and extensions, change file dates, print and save file and folder lists etc.
The program is very useful if - for example - you want to print out an index of the content of a CD or change all file dates to
the current date.
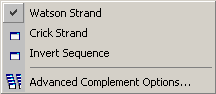
4.2.5 complementing sequences
The Watson / Crick options generates the complementary DNA sequence and displays it with the 5' end
to the left. Invert sequence inverts the current sequence and should be used with caution. The function is useful
when copying sequences written 3' to 5'. In all other cases , i.e. with sequences written 5' to 3' create the inverted
sequence will have no relationship to the original sequence.
Note - The information describing the orientation of the DNA sequence is saved with the file and retrieved when the
file is loaded.
In the sequence lists the following codes are used to indicate the orientation of the sequence: WS - Watson strand,
CS - Crick strand, WI - Inverted Watson strand and CI - Inverted Crick strand. In cases where
orientation information cannot be retrieved or is incomplete, ?'s replaces one or both orientation characters.
The Complement / Invert operation can be performed on the entire project by using the Advanced
Complement Options, a batch version of the above functions.
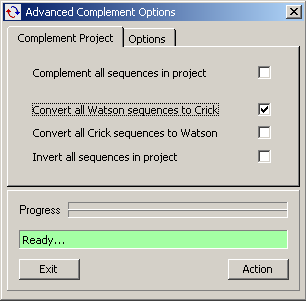
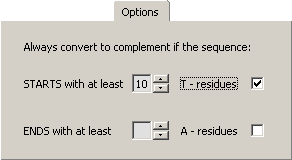
In case you only wish to batch complement polyA sequences set the minimum number of A's / T's for complementation
to be performed.
4.2.6 trim raw sequences
This form includes five utilities for processing raw sequence data. All functions allow you either
to process sequences one-at-a-time in step mode or to launch auto-trimming. While auto-trimming is running
the operation can be paused and the user taking over continuing stepwise. All functions also include undo and reset buttons
letting you reset trimmed sequences contained in the project to the state prior to a trimming operation.
To save resources, you have the option of turning the undo function off before opening the form. In this case,
the undo button is not shown.
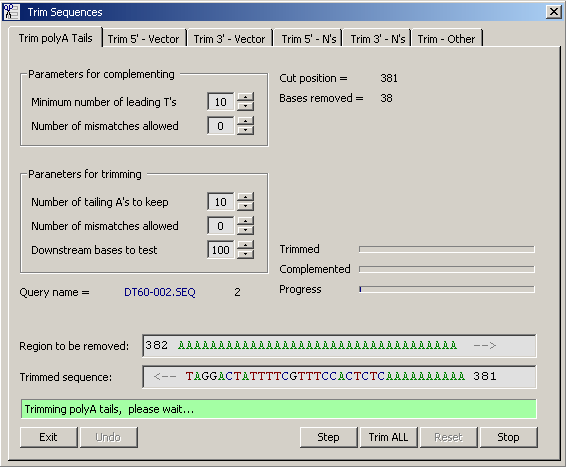
4.2.6.1 Remove PolyA Tails - This function is designed for removing all bases upstream of a leading polyT region. In EST
sequencing from the 3' end all inserts normally contain stretch of T corresponding to the polyA tail of the cDNA clone.
In situations where the sequencing primer position is very close to the start of the insert, the upstream vector part of the
sequence is often biased by dye terminator signals and is not recognised by a comparison with the sequence of the
vector. This function only considers the T's of the and thus trims correctly, also in cases where upstream vector
sequence is ambiguous.
The options and the output of the function is illustrated by the screen dump below. In cases where you wish to
reduce the length of leading T stretches, this can be done by entering the maximum number of T's to retain after
trimming.
4.2.6.2 Remove Vector Sequence - Based on a database containing the sequence of the cloning vector(s) this function
performs a blastn search, evaluates the result and trims the sequence if the selected criteria are met. The matching region and
the start of the sequence after trimming are displayed in the two fields if you use the step option otherwise the main editor
form is hidden to avoid using resources on updating and displaying the sequence. The settings as well as an example of the
output is shown below.
Please note that this function require that a local vector database is already created. Use the functions for creating local
databases if a suitable vector database is not available.

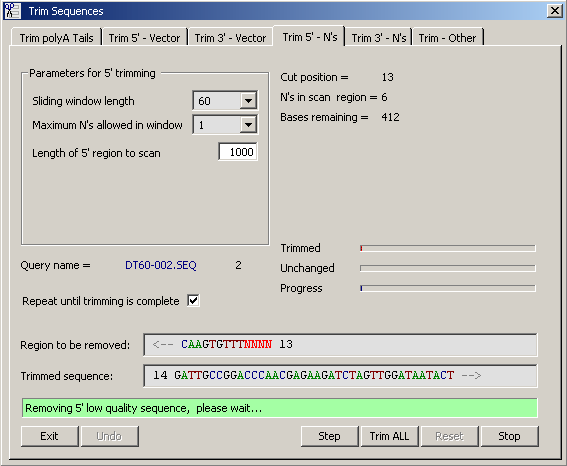
4.2.6.3 Remove Low Quality Sequence - After removal of vector sequence, low quality sequence regions can be
automatically removed from the 5' and 3' ends of the raw sequences. The function determines the number of N's in a window
sliding from the start/end of the sequence. The first time a window-sized region is encountered which meets the selected
criteria, trimming occurs at the most upstream/downstream position of the window. By default trimming is repeated until
all low quality regions are removed.
This function for removing low quality sequence is less accurate - but is much simpler to use - than the function included with
the basecalling facility exploiting the external basecallerLifeTrace.
4.2.6.4 Simple trimming - This function (not illustrated) allows you to either cut the sequences at fixed 5' and 3'
positions or to enter a 5' and 3' string which must exactly match the sequence for trimming to occur. Cutting occurs at
the first position downstream of the 5' string and at the first position upstream of the 3' string. If a perfect match is not
found, no cutting occurs.
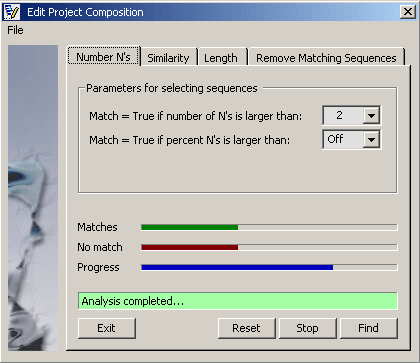
4.2.7 edit project composition
With the functions on this form you can edit the composition of the current project by removing specific
sequence groups such as low quality sequences (with a large number of N's), sequences with significant match to vector
sequences etc.
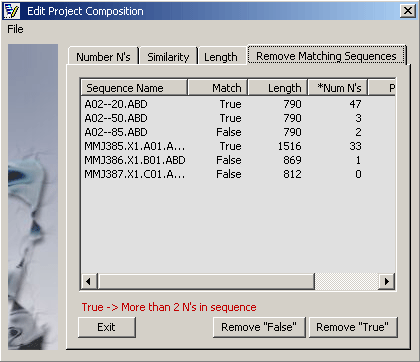
4.2.7.1 Remove low quality sequences - Enter either maximum number or precentage of N' accepted in a sequence and
click the Find command button. The function will analyse the project and display the result in the results
tab, Remove Matching Sequences. Each sequence is labelled True or False indicating whether or
not the specified crieteria were met.
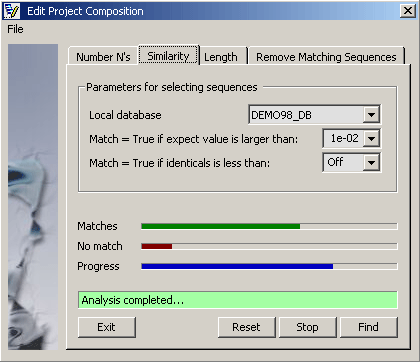
4.2.7.2 Similarity analysis - With this function each sequence in the project is compared to the selected local
database. Running the function with the set parameters then divides the sequences contained in the project into a True
and a False group. Either group can subsequently be removed from the project.
4.2.7.3 Remove short/long sequences - The last function simply measures sequence length and splits the project
sequences into two groups depending on the set length cutoff.
4.2.8 cut / copy / paste
Trivial Windows functions for moving sequences from one instance of SEQtools to another, importing sequences etc. Click Update to format the an imported sequence.
4.2.9 show chromatogram
Viewing and editing chromatograms is performed by the external program. Chromas runs completely independent of
SEQtools except for opening trace files from within SEQtools. Read more about chromatograms and the assiciation between the
project sequence and the chromatogram under theFileandPreferencesmenus.
© 2002-2010S.W. Rasmussen (revised: )